
The standard model of measuring efficacy as a function of receptor occupancy was deemed to be insufficient to adequately explain the dose-response relationships often observed, that were steeper than those predicted by the law of mass action; or why sometimes a supra maximal drug concentration failed to elicit a maximal response within previously observed systems. This was predominantly manifested by a lack of a linear correlation, as predicted by the hyperbolic Langmuir isotherm, between the observed responses of a ligand on its tissues versus the fractions of occupied receptors. In 1956, R. P. Stephenson argued that the magnitude of response, in a tissue, to a given stimulus was the product of the intrinsic efficacy ")

Magnitude of stimulus  = e_A \rho_{AR}")
![S = e_A\frac{[A]}{K_A + [A]}](http://s0.wp.com/latex.php?latex=S+%3D+e_A%5Cfrac%7B%5BA%5D%7D%7BK_A+%2B+%5BA%5D%7D&bg=ffffff&fg=000&s=0 "S = e_A\frac{[A]}{K_A + [A]}")
Where e = efficacy


This definition posits that an agonist with high efficacy would have to occupy only a small fraction of the receptors in order to elicit a maximal response. However, this explanation still failed to take into account the fact that the occupation of the receptors and activation of the receptor are not equivalent events and that the agonist can occupy receptors that are in both the active state or the inactive state. This concept was developed further in the del Castillo-Katz model (Del Castillo and Katz 1957) wherein an isomerization constant was used to define the shift of the receptor between the active and inactive states. This model also showed that total receptor occupancy was not really the limiting factor in determining tissue activation suggesting an existence for the role of receptor reserve in determining the sensitivity of a tissue system to an agonist.
The del Castillo-Katz scheme is based on the following equilibrium relationship wherein the empty receptor (R) isomerizes between the agonist bound inactive state (AR) and the agonist bound active state (AR*):
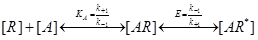 Where the following equilibrium equations can be applied based on the law of mass action:
Where the following equilibrium equations can be applied based on the law of mass action:
![[A] \rho_R = K_A \rho_{AR}](http://s0.wp.com/latex.php?latex=%5BA%5D+%5Crho_R+%3D+K_A+%5Crho_%7BAR%7D&bg=ffffff&fg=000&s=0 "[A] \rho_R = K_A \rho_{AR}")

Where, latex$ K_A$ and E are the respective equilibrium constants as follows:


It also follows from the law of mass action that:

We can thus solve for the fraction of receptors in the AR* state by using equations 1.10 and 1.20 and substituting into equation 1.21 as follows:
![\frac{K_A}{E[A] \rho_{AR^*}} + \frac{1}{E \rho_{AR^*}} + \rho_{AR^*} = 1](http://s0.wp.com/latex.php?latex=%5Cfrac%7BK_A%7D%7BE%5BA%5D+%5Crho_%7BAR%5E%2A%7D%7D+%2B+%5Cfrac%7B1%7D%7BE+%5Crho_%7BAR%5E%2A%7D%7D+%2B+%5Crho_%7BAR%5E%2A%7D+%3D+1&bg=ffffff&fg=000&s=0 "\frac{K_A}{E[A] \rho_{AR^*}} + \frac{1}{E \rho_{AR^*}} + \rho_{AR^*} = 1")
![\rho_{AR^*} = \frac{E[A]}{K_A + (1+E)[A])}](http://s0.wp.com/latex.php?latex=%5Crho_%7BAR%5E%2A%7D+%3D+%5Cfrac%7BE%5BA%5D%7D%7BK_A+%2B+%281%2BE%29%5BA%5D%29%7D&bg=ffffff&fg=000&s=0 "\rho_{AR^*} = \frac{E[A]}{K_A + (1+E)[A])}")
This equation sets itself apart from the Hill-Langmuir isotherm and defines maximal response as a function of 

Substituting 

Or

}{E}")
Substituting in the value of
![\rho_{occupied} = (\frac{E[A]}{K_A+(1+E)[A]})(\frac{1+E}{E})](http://s0.wp.com/latex.php?latex=%5Crho_%7Boccupied%7D+%3D+%28%5Cfrac%7BE%5BA%5D%7D%7BK_A%2B%281%2BE%29%5BA%5D%7D%29%28%5Cfrac%7B1%2BE%7D%7BE%7D%29&bg=ffffff&fg=000&s=0 "\rho_{occupied} = (\frac{E[A]}{K_A+(1+E)[A]})(\frac{1+E}{E})")
Or
}{K_A + (1+E)[A]})](http://s0.wp.com/latex.php?latex=%5Crho_%7Boccupied%7D+%3D+%28%5Cfrac%7B%5BA%5D%281%2BE%29%7D%7BK_A+%2B+%281%2BE%29%5BA%5D%7D%29&bg=ffffff&fg=000&s=0 "\rho_{occupied} = (\frac{[A](1+E)}{K_A + (1+E)[A]})")
This simplifies to,
![\rho_{occupied} = (\frac{[A]}{\frac{K_A}{1+E} + [A]})](http://s0.wp.com/latex.php?latex=%5Crho_%7Boccupied%7D+%3D+%28%5Cfrac%7B%5BA%5D%7D%7B%5Cfrac%7BK_A%7D%7B1%2BE%7D+%2B+%5BA%5D%7D%29&bg=ffffff&fg=000&s=0 "\rho_{occupied} = (\frac{[A]}{\frac{K_A}{1+E} + [A]})")
Where 
![\rho_{occupied} = \frac{[A]}{K_{effective} + [A]}](http://s0.wp.com/latex.php?latex=%5Crho_%7Boccupied%7D+%3D+%5Cfrac%7B%5BA%5D%7D%7BK_%7Beffective%7D+%2B+%5BA%5D%7D&bg=ffffff&fg=000&s=0 "\rho_{occupied} = \frac{[A]}{K_{effective} + [A]}")
Where, 
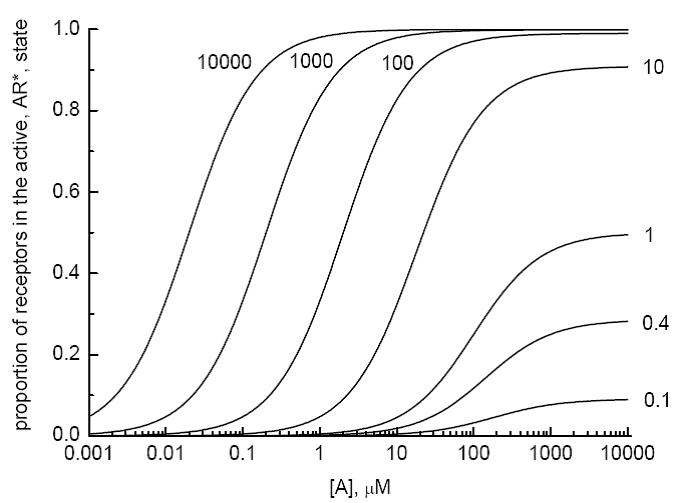
Figure 1. Equation 1.22 predicts a relationship between
Looking at the relationship between the proportions of active receptors to the proportion of occupied receptors as defined in Eq. (1.25), it is easy to see that when an agonist is applied at a very high concentration where all the receptors are fully occupied (i.e. [A] >> Rt and 

